Advanced usage
apytram allows almost full customization by the user. Thus, there are many parameters that can be set and the section below is a bit long!
- Change the number of threads
- Access to temporary files
- Use several input files
- Restart a job
- Add iterations to a job
- Final filter
- Use stranded data
This can be set with the -threads $nb_threads option.
./apytram.py -d $db_name -dt $db_type -fq $fastq_name -q $query_file_name -threads $nb_threads
By default, it is equal to 1.
If you use the -tmp option, temporary files will not be deleted at the end of the job.
./apytram.py -d $db_name -dt $db_type -fq $fastq_name -q $query_file_name -tmp $tmp_directory_name
You can give several space delimited file names to apytram with the option -fa and -fq.
The options -fa and -fq can not be give in a same time.
- For several input files (
$fastq1 $fastq2 ...) in fastq format containing the reads:
apytram.py --database $db_name -dt $db_type -fq $fastq1 $fastq2 -q $query_file_name
- For several input file (
$fasta1 $fasta2 ...) in fasta format containing the reads:
apytram.py --database $db_name -dt $db_type -fa $fasta1 $fasta2 -q $query_file_name
If you want to restart a job IN CASE OR FAIL OR WHATEVER, you can launch your command with the same temporary directory (-tmp option ). In this case, apytram will see files have ever been generated and will not loose time to execute again time-consuming sub-commands as Trinity or Blast. apytram will begin again to work when it will not find a given temporary file.
If you want to continue iteration for a job, you can use the -i_start option only if the -tmp option was set for the job you want to restart.
./apytram.py -d $db_name -dt $db_type -fq $fastq_name -q $query_file_name -tmp $tmp_directory_name -i_start X
apytram will begin new iteration from temporary files of the Xth iteration.
In case you would only like to build a BLAST database out of your data, you can simply do this within apytram, omitting the -q option.
./apytram.py -d $db_name -t $db_type -fq $fastq_name
A final filter (using the -fmal, -fid and -flen options) can be applied on the reconstructed sequences to be more stringent than the threshold used during the iterative process.
The reference sequence of a reconstructed sequence is the sequence from the query file that is the more homologous to it (according to Exonerate score, see "How does apytram work?").
If there are several references, the filter will be different for each reference. For example, if there are a reference A and a reference B. Reconstructed sequences more homologous to A will be filtered according a filter calculated from A and reconstructed sequences more homologous to B will be filtered according a filter calculated from B.
-
If you want to keep only reconstructed sequences that have a length superior to X percent of the reference sequence:
./apytram.py -d $db_name -t $db_type -fq $fastq_name -q $query_file_name -flen X -
If you want to keep only reconstructed sequences that have an identity percentage superior to Y percent with the reference sequence on the whole alignment length:
./apytram.py -d $db_name -t $db_type -fq $fastq_name -q $query_file_name -fid Y -
If you want to keep only reconstructed sequences that align on the reference on a length superior to Z percent of the reference sequence length: ``` ./apytram.py -d $db_name -t $db_type -fq $fastq_name -q $query_file_name -fmal Z
```
- If you want to combine all these options, it's possible:
./apytram.py -d $db_name -t $db_type -fq $fastq_name -q $query_file_name -flen X -fid Y -fmal Z
This can be set with the -dt option.
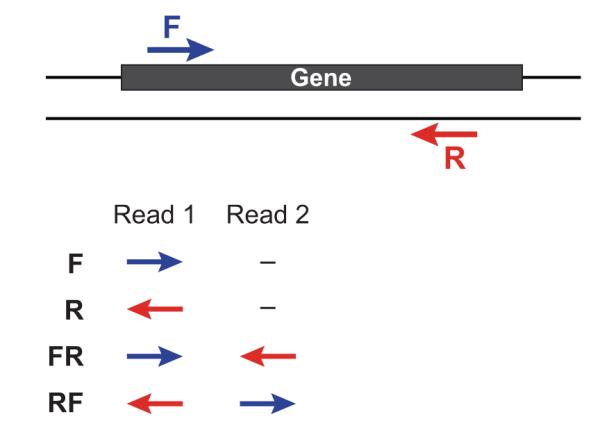
If you have stranded data, you can use RF, FR, F or R to specify your library type as defined by the Trinity documentation. Extract of the Trinity documentation:
If you have strand-specific data, specify the library type. There are four library types:
Paired reads:
RF: first read (/1) of fragment pair is sequenced as anti-sense (reverse(R)), and second read (/2) is in the sense strand (forward(F)); typical of the dUTP/UDG sequencing method.
FR: first read (/1) of fragment pair is sequenced as sense (forward), and second read (/2) is in the antisense strand (reverse)
Unpaired (single) reads:
F: the single read is in the sense (forward) orientation
R: the single read is in the antisense (reverse) orientation