Home
- Preamble
- Quick start
- Installing apytram
- How to use apytram?
- How does apytram work?
- How to get help?
- Citing
This software is inspired from aTRAM (see References) but was implemented in Python and internal strategies have been designed differently. In particular, several species can be processed together.
Any question or suggestion on the program can be addressed to: carine.rey@ens-lyon.fr
apytram allows assembling sequences from RNA-Seq data (paired-end, single-end, stranded or not) using one or more guide sequences. The guide sequences can come from the species of interest or from a different species.
To install apytram you need:
Or get a test version of apytram with almost no instalation:
- Get the apytram docker container; more information --> here
For impatient users or preliminary tests, here are some command examples. For more information, see other example usage or Advanced usage:
- For a single input species with a file (
$fastq_filename) in fastq format containing the reads:
apytram.py -d $db_name -dt $db_type -fq $fastq_filename -q $query_file_name
- For a single input species with a file (
$fasta_filename) in fasta format containing the reads:
apytram.py -d $db_name -dt $db_type -fa $fasta_filename -q $query_file_name
- For several species using fastq formated files:
apytram.py -d $db_name1:SP1,$db_name2:SP2 -dt $db_type1:SP1,$db_type2:SP2 -fq $fastq_filename1:SP1,$fastq_filename2:SP2 -q $query_file_name
- For several species using fasta formated files:
apytram.py -d $db_name1:SP1,$db_name2:SP2 -dt $db_type1:SP1,$db_type2:SP2 -fq $fasta_filename1:SP1,$fasta_filename2:SP2 -q $query_file_name
- For several species using fasta formated files and several query filenames:
apytram.py -d $db_name1:SP1,$db_name2:SP2 -dt $db_type1:SP1,$db_type2:SP2 -fq $fasta_filename1:SP1,$fasta_filename2:SP2 -q $query_filename1:Q1,$query_filename2:Q2
with:
-
$db_typebeing "single" or "paired" for un-stranded data or "RF","FR","R" or "F" for stranded data (see here for more details) -
$query_filenameis a fasta file containing one or more baits that will be used to capture reads before assembly. See below for more details. -
$db_nameis the localization of a BLAST formated read database which will be created by apytram if it do not exist.
The run will look like:
user@linux:$ apytram.py --database $db_name -dt $db_type -fq $fastq_name -q $query_file_name
[Running in process...]
[Warning messages may appear, but they are not errors. If real errors appear, the process will stop]
[...]
user@linux:$ and the output of this command will be 3 files (More options on the output files are specified in the Output files section):
-
A fasta file containing all the assembled sequences
-
A fasta file containing the best assembled sequences
-
A log file containing run information
N.B.: The query file must look like this:
>query_sequence1
ATGATATATATTAGAGGAGAGACAGCAGCAGTACTATGA
>query_sequence2
ATGATATATATTAGAGGAGAGACAGCAGCAGTATGA
It can contain several sequences but they must be homologous because they will be treated together in a unique run. They can be for instance different transcript sequences for the same gene, incomplete coding sequences...
If one of your reference contained a shared domain, all genes which shared this domain will be recruited. It is a logical consequences but it will increase considerably execution time of apytram. To avoid huge runs of apytram, a time limit (2 hours) has been set up. This limit can be increased. (see Advanced usage)
Installing apytram on Linux has to be done in two steps. As apytram uses different external software, you have to install those dependencies and only then install apytram itself.
apytram is made for command-line usage. It has been tested on Linux and Mac machine:
- Debian/Ubuntu users:
This installation wiki is made for Debian/Ubuntu users. Commands can just be copied and pasted in your favorite terminal.
- MacOS and CentOS users:
As this installation wiki is made for Debian/Ubuntu users, command line must be adapted. In fact, there are only package management program which must be change according to your OS (yum, macports, ...).
apytram is written in python 2.
apytram is available in a docker container available in DockerHub.
(If you don't have docker, you can find here installation instruction.)
You can get and run the container with this unique command:
docker run -t -i -v $your_data:/data carinerey/apytramAnd you're done!
The -v option allows you to share a directory ($your_data) of your computer with the virtual environment in the docker container.
apytram can be called directly in the docker container terminal.
Warning, the container is in development, if you have any problem don't hesitate to contact me (carine.rey@ens-lyon.fr).
You can get apytram by:
- By downloading source code:
apytram is available on GitHub https://github.com/CarineRey/apytram. Click on the "Download ZIP" button once on that page.
Then, extract the file where you want.
- By command line:
This way is much faster. The command below will create in your current directory a directory named "apytram":
git clone https://github.com/CarineRey/apytram.gitAnd you're done!
(If you don't have git, you can find here installation instruction.)
You can easily check if all dependencies are satisfied or which have to be installed by executing the test_apytram_configuration.py script which is located in the apytram directory.
In the apytram directory type:
./test_apytram_configuration.pyor
make testIf you want to move apytram.py to another directory, don't forget to bring with it its libraries (the ApytramLib directory) or add the path of the ApytramLib directory in the PYTHONPATH.
For instance:
export PYTHONPATH=/path/to/ApytramLib:$PYTHONPATHTo install Python modules, you can use pip command and install it by typing: sudo apt-get install python-pip. Then, install the 2 modules:
-
pandas: type
sudo pip install pandas -
Matplotlib >= 1.13: type
sudo pip install Matplotlib -
Biopython : type
sudo pip install biopython
Note that the programs must be available in the $PATH environment variable. To add a program to the $PATH environment variable, type in your terminal:
export PATH=/path/to/the/new/program/:$PATHYou can install these programs either by manual installation of each of them in following links or via a package management program when available but you must verify the version.
-
Trinity >= v2.3 (with its dependencies: Samtools = v0.1.19-1, Java 1.8 and Bowtie2)
-
Download Trinity here (note that you have to set the path to the folder containing the Trinity executable, and not to the executable itself; for example
PATH=/path/to/trinityrnaseq-2.3.0/:$PATH) -
sudo apt-get install samtools=0.1.19-1or Download Samtools here -
sudo apt-get install bowtie2or Download Bowtie here
-
-
Mafft >= v7
sudo apt-get install mafftor Download Mafft here -
Seqtk >= 1.2
Toolkit for processing sequences in FASTA/Q formats https://github.com/lh3/seqtk
``` sh
mkdir -p /home/user/bin/seqtk
git clone https://github.com/lh3/seqtk.git /home/user/bin/seqtk
cd /home/user/bin/seqtk
make
```
- BLAST+ 2.2.28-2 or 2.6.0
```sudo apt-get install ncbi-blast+=2.2.28-2``` or <a href="http://www.ncbi.nlm.nih.gov/books/NBK52640/#chapter1.Downloading" target="_blank">Download BLAST+ here</a>
- TransDecoder 5.0.2
``` sh
mkdir -p /home/user/bin/TransDecoder
transdecoder_version=TransDecoder-v5.0.2
cd /home/user/bin/TransDecoder
wget https://github.com/TransDecoder/TransDecoder/archive/"$transdecoder_version".zip &&\
unzip "$transdecoder_version".zip && rm "$transdecoder_version".zip && \
&& cd /home/user/bin/TransDecoder/TransDecoder-"$transdecoder_version"/ && make
PATH=/home/user/bin/TransDecoder/TransDecoder-"$transdecoder_version"/:$PATH
```
This example is included in the apytram directory using a nucleotide query. It should run in about a dozen seconds. Code must be executed in the parent directory of apytram. Before running this example, check that all dependencies are verified by executing the test_configuration.py script.
cd /path/to/apytram/parent/directory/
export OUT="exec_example"
apytram/test_configuration.py
apytram/apytram.py -d $OUT/db/examplefq \
-dt paired \
-fq apytram/example/example_db.fastq \
-q apytram/example/ref_gene.fasta \
-out $OUT/apytram \
-log $OUT/apytram.log \
--plot \
--plot_ali In this example, apytram creates a new folder in your working directory named exec_example. This folder contains several files, as specified by the command above.
user@bla:~/path/to/apytram/exec_example/ ls -1
apytram.ali.fasta
apytram.ali.png
apytram.best.fasta
apytram.fasta
apytram.log
apytram.stats.csv
apytram.stats.pdf
db/Note that the /exec_example/db/ folder contains the database generated by apytram.
Run information is in the apytram.log file. This file contains details on the running process, i.e. details on the processing time of all the programs implemented in apytram (Trinity, BLAST, Mafft, etc.), the iteration number and the time taken by each iteration (in this example, there is only one).
-
apytram.fasta:A fasta file containing all reconstructed sequences of the last iteration that pass the final filter.
-
apytram.best.fasta:A fasta file containing the reconstructed sequences of the last iteration that have the best homology score according to exonerate, and that pass the final filter.
-
apytram.stats.pdf:
Available if the ```--plot``` option is set. This produces a *.pdf* file containing 2 figures containing global information at each iteration, such as the evolution of the number of contigs after each iteration, iteration time, best and average identity with the reference, time taken by each program in apytram (such as Trinity, ...), etc. There are many figures that can be useful if you want to "know what happens" at each iteration.
-
apytram.stats.csv:
Available with the ```--plot``` or ```--stats``` options. This produces a *.csv* file containing the raw data used to draw the figures present in the *.stats.pdf* file
-
apytram.ali.png:Available if the
--plot_alioption is set. This produces a figure representing the alignment of all reconstructed sequences with the query (only contigs that pass the final filter are shown). In the example shown here, there is only one contig. White represents a gap, blue a base of the reference, green an identical base of the reference in a reconstructed sequence, red a different base compared with the reference and yellow a base corresponding to a gap in the reference. -
apytram.ali.fasta:Alignment that is used to generate the apytram**.ali.png** file.
apytram allows almost full customization by the user. Thus, there are many parameters that can be set and the section below is a bit long!
- Change the number of threads
- Access to temporary files
- Use several species
- Use several queries
- Restart a job
- Add iterations to a job
- Final filter
- Use stranded data
- Speed optimization
- Optimizing the accuracy
- Following the program's progress
This can be set with the -threads $nb_threads option.
./apytram.py -d $db_name -dt $db_type -fq $fastq_name -q $query_file_name -threads $nb_threadsBy default, it is equal to 1.
If you use the -tmp option, temporary files will not be deleted at the end of the job.
./apytram.py -d $db_name -dt $db_type -fq $fastq_name -q $query_file_name -tmp $tmp_directory_nameYou can give several coma delimited file names to apytram with the option -fa and -fq.
The options -fa and -fq can not be give in a same time.
- For several species using fastq formated files:
apytram.py -d $db_name1:SP1,$db_name2:SP2 -dt $db_type1:SP1,$db_type2:SP2 -fq $fastq_filename1:SP1,$fastq_filename2:SP2 -q $query_file_name:Q1 -o $prefix- For several species using fasta formated files:
apytram.py -d $db_name1:SP1,$db_name2:SP2 -dt $db_type1:SP1,$db_type2:SP2 -fq $fasta_filename1:SP1,$fasta_filename2:SP2 -q $query_file_name:Q1 -o $prefixBy default, all output sequences will be put in the same outputfile, $prefix.Q1.fasta, you can use the --output_by_species to build an output file by species, $prefix.Q1.SP1.fasta, $prefix.Q1.SP2.fasta.
You can give several coma delimited file names to apytram with the option -q.
You must specified each query name to build an output for each query.
- For several queries:
apytram.py -d $db_name:SP1 -dt $db_type:SP1 -fq $fastq_filename:SP1 -q $query_filename1:Q1,$query_filename2:Q2If you want to restart a job IN CASE OR FAIL OR WHATEVER, you can launch your command with the same temporary directory (-tmp option ). In this case, apytram will see files have ever been generated and will not loose time to execute again time-consuming sub-commands as Trinity or Blast. apytram will begin again to work when it will not find a given temporary file.
If you want to continue iteration for a job, you can use the -i_start option only if the -tmp and --keep_tmp options were set for the job you want to restart.
./apytram.py -d $db_name -dt $db_type -fq $fastq_name -q $query_file_name -tmp $tmp_directory_name --keep_tmp -i_start Xapytram will begin new iteration from temporary files of the Xth iteration.
In case you would only like to build a BLAST database out of your data, you can simply do this within apytram, omitting the -q option.
./apytram.py -d $db_name -t $db_type -fq $fastq_nameA final filter (using the -fmal, -fid and -flen options) can be applied on the reconstructed sequences to be more stringent than the threshold used during the iterative process.
The reference sequence of a reconstructed sequence is the sequence from the query file that is the more homologous to it (according to Exonerate score, see "How does apytram work?").
If there are several references, the filter will be different for each reference. For example, if there are a reference A and a reference B. Reconstructed sequences more homologous to A will be filtered according a filter calculated from A and reconstructed sequences more homologous to B will be filtered according a filter calculated from B.
-
If you want to keep only reconstructed sequences that have a length superior to X percent of the reference sequence:
sh ./apytram.py -d $db_name -t $db_type -fq $fastq_name -q $query_file_name -flen X -
If you want to keep only reconstructed sequences that have an identity percentage superior to Y percent with the reference sequence on the whole alignment length:
sh ./apytram.py -d $db_name -t $db_type -fq $fastq_name -q $query_file_name -fid Y -
If you want to keep only reconstructed sequences that align on the reference on a length superior to Z percent of the reference sequence length: ```sh ./apytram.py -d $db_name -t $db_type -fq $fastq_name -q $query_file_name -fmal Z
```
- If you want to combine all these options, it's possible:
./apytram.py -d $db_name -t $db_type -fq $fastq_name -q $query_file_name -flen X -fid Y -fmal Z
This can be set with the -dt option.
If you have stranded data, you can use RF, FR, F or R to specify your library type as defined by the Trinity documentation. Extract of the Trinity documentation:
If you have strand-specific data, specify the library type. There are four library types:
Paired reads:
RF: first read (/1) of fragment pair is sequenced as anti-sense (reverse(R)), and second read (/2) is in the sense strand (forward(F)); typical of the dUTP/UDG sequencing method.
FR: first read (/1) of fragment pair is sequenced as sense (forward), and second read (/2) is in the antisense strand (reverse)
Unpaired (single) reads:
F: the single read is in the sense (forward) orientation
R: the single read is in the antisense (reverse) orientation
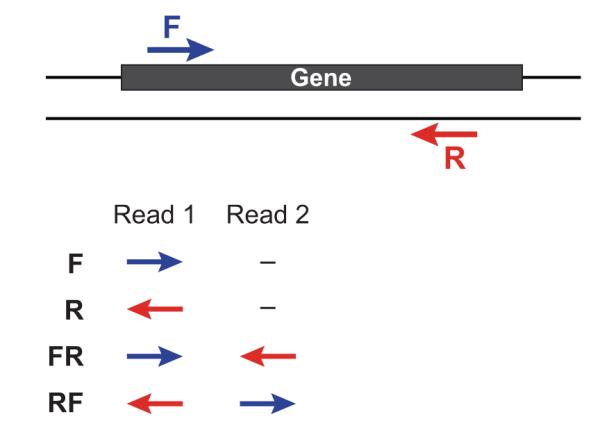
Paired-end RNA-seq data run faster than single-end data.
To save time:
-
reduce evalue (-e) -
not use -tmp option -
not use --keep_iterations -
not use --finish_all_iter -
not use the --plot and --plot_ali options
If you want to use apytram on several query files and you have several available threads, it is more efficient to minimize the number of threads by apytram job than to maximize the number of threads by job. Each job will be slower but at the end you will save time. This is due to the non linearity of the time saved by Trinity and Blast when the number of threads is increased.
See folowing options:
- -e
- -mal
- -id
- -len
- -fid
- -fmal
- -flen
A job can take some minutes to several hours to complete. To know the progress of your job you can look into the log file (-log option or by default apytram.log).
You can look at the $OUTPUT_PREFIX.stats.pdf (--plot option) at the end the job, to have general information on the progress of your job. All values needed to create the plot in $OUTPUT_PREFFIX.stats.pdf are available in $OUTPUT_PREFFIX.stats.csv. The $OUTPUT_PREFFIX.stats.csv file can be only created using the --stats option.
The name of the log file containing run information can be changed using the -log option.
./apytram.py -d $db_name -dt $db_type -fq $fastq_name -q $query_file_name -log $LOGAs a result of an apytram run, several output files are written in your working directory. You can specify their name using the -out option. For instance:
./apytram.py -d $db_name -dt $db_type -fq $fastq_name -q $query_file_name -out $OUTPUT_PREFIX-
$OUTPUT_PREFIX.fasta:A fasta file containing all reconstructed sequences of the last iteration that pass the final filter.
-
$OUTPUT_PREFIX.best.fasta:A fasta file containing the reconstructed sequences of the last iteration with the best homology scores with the bait sequence (according to exonerate), and that pass the final filter.
-
$OUTPUT_PREFIX.stats.pdf:
Available if the ```--plot``` option is set. This produces a *.pdf* file containing 2 figures containing global information at each iteration.
-
$OUTPUT_PREFIX.stats.csv:
Available with the ```--plot``` or ```--stats``` options. This produces a *.csv* file containing the raw data used to draw the figures present in the *.stats.pdf* file
-
$OUTPUT_PREFIX.ali.png:A figure representing the alignment of all reconstructed sequences with the query (only contigs that pass the final filter are shown). White represents a gap, blue a base of the reference, green an identical base of the reference in a reconstructed sequence, red a different base compared with the reference and yellow a base corresponding to a gap in the reference.
-
$OUTPUT_PREFIX.ali.fasta:Alignment that is used to generate the
$OUTPUT_PREFIX.ali.png file.
As apytram runs, it generates temporary files at each iteration that are finally removed, so that the user only have the final result. If ever you would be interested in looking into more detail at what happens at each iteration, you might want to keep these files using the -tmp /path/to/my/temporary/files/ option.
With the --keep_iterations option, a fasta file containing the reconstructed sequences after each iteration will be created.

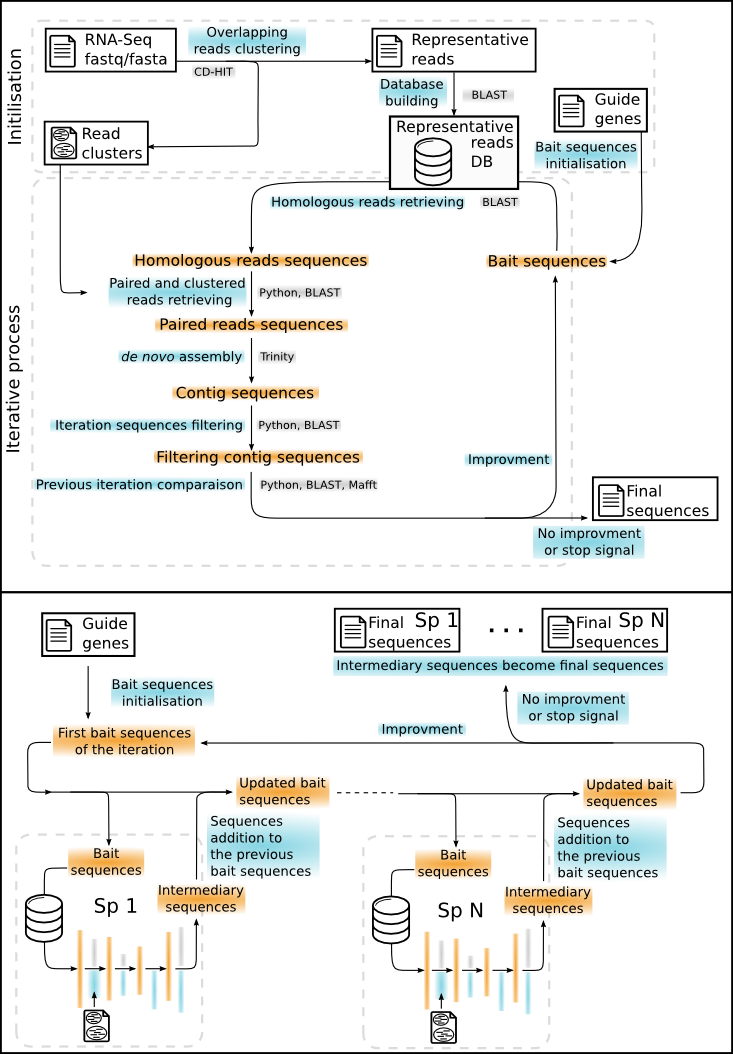
apytram allows the assembly of sequences from RNA-seq data (paired-end or single-end) using one or more reference homologous sequences. The reference sequences can come from the species of interest or from a different species.
For this, the program will need:
- a database: usually your RNA-Seq reads, formatted as a BLAST database (see Database building section)
- a query file in fasta format, with the homologous sequence(s) of your gene(s) of interest
A basic run consists in (see Iterative process section for more detail):
-
aligning the query sequence(s) on the database
-
keeping the reads that align with the query sequence(s)
-
assembling those reads into contigs
-
using the contigs as the new reference for the next iteration (perform steps 1 to 3 with this new reference: obtention of longer contigs)
-
comparing contigs from iteration i and iteration i-1
-
deciding if the assembly is better and if it's worth starting a new iteration (see the Criteria to stop the iterative process section)
The RNA-seq data (a fastq or fasta file), given by the -fq or -fa option, is formatted into a BLAST database, whose name is given by the -d option.
The data type, paired or single end, must be given by the -dt option. If data is paired-end, all reads (1 and 2) must be concatenated in a single file. WARNING: Paired read names must end with 1 or 2.
Reads contained in this file will all be used, so the file must have already been cleaned up.
Note that if your data is in fastq format, it will be converted into a fasta file. This conversion can take some minutes.
As the database building step is time consuming, if a BLAST database is already present, the database building will be skipped.
Sequences in the query file (-q option) will serve as the first reference sequences. The possibility to use a multi-reference query file is still in testing.
-
Reads recruting (BLAST):
Bait sequences are used to recrute homologous reads by BLAST. The
-eoption allows fixing the evalue threshold (default: 0.001). If the data is paired-end, all paired reads are added to the read list.Names (.txt format) and sequences (.fasta format) of all these reads are accessible in temporary files if the
-tmpoption is used. -
Reads assembly (Trinity):
All reads found by BLAST (present in the temporary file) are assembled de novo by Trinity with default parameters. Note that If the data contains paired-end reads, Trinity takes reads as paired reads.
-
Quality and filtering of reconstructed contigs (Exonerate or Blast (faster)):
Exonerate is used to align each assembled contig to the reference. The length of the contig, the length of the alignment with the reference, the percentage of identity and the alignment score are collected. Reconstructed sequences are filtered according to the options
-id,-maland-len. Contigs that pass all filters are used as reference for the next iteration. -
Comparison with previous iteration (Exonerate or Blast (faster)):
Exonerate is used to compare the reconstructed sequences of iteration n+1 with iteration n. If the quality of the assembled seqeunce is not improved, the process is stopped.
-
Coverage calculation (Mafft):
So as to estimate the overall quality of the contig assembly, apytram calculates 2 coverage values:
- a Strictcoverage that represents the percentage of the query sites with a homologous site in the reconstructed sequence(s)
- a Largecoverage that represents the percentage of sites in the alignment with at least one representative in the reconstructed sequence(s) divided by the length of the reference so by definition it can be superior to 100. Note that if there is not only one reference sequence, the first sequence in the reference file is took as reference for the coverage counter but all references are aligned.

At the end of an iteration, reconstructed sequence(s) become the reference sequences for the next iteration.
If one of these criteria is completed during an iteration, the iterative process will stop. The following criteria are implemented as default settings :
- The number of max iteration (
-ioption) is reached. - Recruted reads are the same as in the previous iteration.
- Reconstructed sequences at the end of an iteration are almost the same as after the previous iteration. Almost means each sequence of an iteration has a corresponding sequence in the previous iteration with at least 99% identity and 98% of its length. These threshold are by default and can not be changed. This criteria allows stopping iterations when Trinity assemblies differ of few bases.
- The number of reconstructed sequences has not changed AND the total length, score and LargeCoverage of all reconstructed sequences have not been improved. The use of the Largecoverage value in this step allows to keep iterating if the UTR in 5' and 3' are getting longer, even if the coding sequence does not.
If the --required_coverage option is used, the iterating process will stop if the Strictcoverage is superior to the Required_coverage.
N.B.:None of these criteria is applied if the --finish_all_iter option is used.
The last criteria which can stop the iterative process is a time limit given by the -time_max option (by default 7200 seconds, but this can be changed).
If this time limit is reached, no new iteration will begin even with the --finish_all_iteration option. Please note that this means that a job can thus spend more than the -time_max limit if the database building and the last iteration last more than -time_max setting.
A final filter (-fmal, -fid and -flen options) can be applied on the reconstructed sequences to be more stringent than the threshold used during the iterative process. See the Advanced usage page for more information.
Finally, several output files are written in your working directory:
-
$OUTPUT_PREFIX.fasta:A fasta file containing all reconstructed sequences of the last iteration that pass the final filter.
-
$OUTPUT_PREFIX.best.fasta:A fasta file containing the best reconstructed sequences of the last iteration for each references of the query file. Thes sequences must have pass the final filter. The best sequence is determined by the best homology score calculated by Exonerate.
-
$OUTPUT_PREFIX.stats.pdf:
Available if the ```--plot``` option is set. This produces a *.pdf* file containing 2 figures containing global information at each iteration.
-
$OUTPUT_PREFIX.stats.csv:
Available with the ```--plot``` or ```--stats``` options. This produces a *.csv* file containing the raw data used to draw the figures present in the *.stats.pdf* file
-
$OUTPUT_PREFIX.ali.png:A figure representing the alignment of all reconstructed sequences with the query (only contigs that pass the final filter are shown). White represents a gap, blue a base of the reference, green an identical base of the reference in a reconstructed sequence, red a different base compared with the reference and yellow a base corresponding to a gap in the reference.
-
$OUTPUT_PREFIX.ali.fasta:Alignment that is used to generate the
$OUTPUT_PREFIX.ali.png file.
apytram.py -hThe help message provides information on all the possible options.
usage: apytram.py [-h] [--version] -d DATABASE -dt DATABASE_TYPE -out
OUTPUT_PREFIX [-fa FASTA] [-fq FASTQ] [-q QUERY]
[-i ITERATION_MAX] [-i_start ITERATION_START] [-log LOG]
[-tmp TMP] [--keep_tmp] [--no_best_file] [--only_best_file]
[--stats] [--plot] [--plot_ali] [-e EVALUE] [-id MIN_ID]
[-mal MIN_ALI_LEN] [-len MIN_LEN]
[-required_coverage REQUIRED_COVERAGE] [--finish_all_iter]
[-flen FINAL_MIN_LEN] [-fid FINAL_MIN_ID]
[-fmal FINAL_MIN_ALI_LEN] [-threads THREADS]
[-memory MEMORY] [-time_max TIME_MAX] [--write_even_empty]
[--out_by_species] [--debug]
Run apytram.py on a fastq file to retrieve homologous sequences of bait
sequences.
optional arguments:
-h, --help show this help message and exit
--version show program's version number and exit
Required arguments:
-d DATABASE, --database DATABASE
Database prefix name. If a database with the same name
already exists, the existing database will be kept and
the database will NOT be rebuilt.
-dt DATABASE_TYPE, --database_type DATABASE_TYPE
single: single unstranded data ______________________
paired: paired unstranded data ______________________
RF: paired stranded data (/1 = reverse ; /2 = forward)
FR: paired stranded data (/1 = forward ; /2 = reverse)
F: single stranded data (reads = forward) ____________
R: single stranded data (reads = reverse) ____________
WARNING: Paired read names must finished by 1 or 2
-out OUTPUT_PREFIX, --output_prefix OUTPUT_PREFIX
Output prefix
Input Files:
-fa FASTA, --fasta FASTA
Fasta formated RNA-seq data to build the database of
reads (only one file).
-fq FASTQ, --fastq FASTQ
Fastq formated RNA-seq data to build the database of
reads (several space delimited fastq file names are
allowed). For paired data, fq must be previously
concatenated. WARNING: Paired read names must finished
by 1 or 2. (fastq files will be first converted to a
fasta file. This process can require some time.)
Query File:
-q QUERY, --query QUERY
Fasta file (nucl) with homologous bait sequences which
will be treated together for the apytram run. If no
query is submitted, the program will just build the
database. WARNING: Sequences must not contain "- * . "
Number of iterations:
-i ITERATION_MAX, --iteration_max ITERATION_MAX
Maximum number of iterations. (Default 5)
-i_start ITERATION_START, --iteration_start ITERATION_START
Number of the first iteration. If different of 1, the
tmp option must be used. (Default: 1)
Output Files:
-log LOG a log file to report avancement (default: apytram.log)
-tmp TMP Directory to stock all intermediary files for the
apytram run. (default: a directory in /tmp which will
be removed at the end)
--keep_tmp By default, the temporary directory will be remove.
--no_best_file By default, a fasta file (Outprefix.best.fasta)
containing only the best sequence is created. If this
option is used, it will NOT be created.
--only_best_file By default, a fasta file (Outprefix.fasta) containing
all sequences from the last iteration is created. If
this option is used, it will NOT be created.
--stats Create files with statistics on each iteration.
(default: False)
--plot Create plots to represent the statistics on each
iteration. (default: False)
--plot_ali Create file with a plot representing the alignement of
all sequences from the last iteration on the query
sequence. Take some seconds. (default: False)
Thresholds for EACH ITERATION:
-e EVALUE, --evalue EVALUE
Evalue threshold of the blastn of the bait queries on
the database of reads. (Default 1e-5)
-id MIN_ID, --min_id MIN_ID
Minimum identity percentage of a sequence with a query
on the length of their alignment so that the sequence
is kept at the end of a iteration (Default 50)
-mal MIN_ALI_LEN, --min_ali_len MIN_ALI_LEN
Minimum alignment length of a sequence on a query to
be kept at the end of a iteration (Default 180)
-len MIN_LEN, --min_len MIN_LEN
Minimum length to keep a sequence at the end of a
iteration. (Default 200)
Criteria to stop iteration:
-required_coverage REQUIRED_COVERAGE
Required coverage of a bait sequence to stop iteration
(Default: No threshold)
--finish_all_iter By default, iterations are stop if there is no
improvment, if this option is used apytram will finish
all iteration (-i).
Thresholds for Final output files:
-flen FINAL_MIN_LEN, --final_min_len FINAL_MIN_LEN
Minimum PERCENTAGE of the query length to keep a
sequence at the end of the run. (Default: 0)
-fid FINAL_MIN_ID, --final_min_id FINAL_MIN_ID
Minimum identity PERCENTAGE of a sequence with a query
on the length of their alignment so that the sequence
is kept at the end of the run (Default 0)
-fmal FINAL_MIN_ALI_LEN, --final_min_ali_len FINAL_MIN_ALI_LEN
Alignment length between a sequence and a query must
be at least this PERCENTAGE of the query length to
keep this sequence at the end of the run. (Default: 0)
Miscellaneous options:
-threads THREADS Number of available threads. (Default 1)
-memory MEMORY Memory available for the assembly in Giga. (Default 1)
-time_max TIME_MAX Do not begin a new iteration if the job duration (in
seconds) has exceed this threshold. (Default 7200)
--write_even_empty Write output fasta files, even if they must be empty.
(Default: False)
--out_by_species Write output fasta files for each species. (Default:
False)
--debug debug mode, default False
Allen, JM, DI Huang, QC Cronk, KP Johnson. aTRAM automated target restricted assembly method a fast method for assembling loci across divergent taxa from next-generation sequencing data. (2015) BMC Bioinformatics Full-text link
Please refer to the Zenodo link below for citation information.
Rey Carine, Lorin Thibault, Sémon Marie, & Boussau Bastien. (2017, June 8). CarineRey/apytram: apytram v1.1. Zenodo. http://doi.org/10.5281/zenodo.804416
- make a pypi distributable package (see https://pypi.python.org/pypi)