Residuals represent the difference between the data and the model and are essential to explore the adequacy of the model.
In the Gaussian case (for the SLR and MLR), the residuals are
These are called response residuals for GLMs but they are not as informative. While before, due to our assumptions, we were expecting homoskedasticity, here heteroskedasticity is not a surprise. In fact say we model a GLM with a Poisson distribution: the variance itself is based on the expected value, which is in turn not constant.
- What we can do is to try and verify whether the proportion between the variance and the expected values stays the same.
- To use the response residuals we need to perform some modification.
We need to remind that analyzing the residuals is more difficult with the GLM since its less clear how to define them and their interpretation.
The Pearson residuals are comparable to the standardized residuals used in linear models and is defined as:
- They should be more or less standardized, namely
$\stackrel{approx}\thicksim \mathcal{N}(0,1)$
$$r^{P}{i} = \frac{y{i}-\hat{y}{i}}{\sqrt{\hat{V}{i}}}$$
residuals(model1,"pearson")[1:5]
1 2 3
2.0428979 -1.2425388 4.0582724
4 5
-1.4815094 0.6455594
We can obtain the deviance residuals as:
residuals(model1)[1:5]
1 2 3
1.7903699 -1.7572153 3.1414336
4 5
-2.0951707 0.6101256These are the default choice of residuals.
If we use
model1$residuals[1:5]
1 2 3
1.0287693 -1.0000000 2.5481128
4 5
-1.0000000 0.3790497we obtain the working residuals, i.e.
$$\hat{\eta}{i} + (y{i} - \hat{\mu}{i})\frac{d\eta{i}}{d\mu_{i}}$$
For ordinary linear models, the plot of residuals against fitted values is probably the single most valuable graphic for model diagnostics.
In GLMs residual plots are much harder to read and verifying assumptions is more complicated (so we don’t really focus on it during the course. For GLMs, we must decide on the appropriate scale for the fitted values.
- Usually, it is better to plot the fitted linear predictors, rather than the predicted responses
$\hat{y}_{i}$
$$\hat{\beta}{0} + \hat{\beta}{1}x_{i,1} + ... + \hat{\beta}{p-1}x{i,p-1}$$
- Scatter plot of Deviance residuals against xj
- Used to check whether any systematic relationship is present: if so include xjin the model.
- Plot working residuals against linear predictor
- Plot should be linear
- If not the link function might be not correctly specified.
- Influential and leverage points can still be problematic: we can define leverage-aware residuals
- We don’t discuss them in the course
For 30 Galapagos Islands, we have a count of the number of plant species found on each island and the number that are endemic to that island. We also have five geographic variables for each island.
We model the number of species using normal linear regression:
data(gala, package="faraway")
gala <- gala[,-2]We throw out the Endemics variable (which falls in the second column of the dataframe) since we won’t be using it in this analysis. We fit a linear regression and look at the residual vs. fitted plot:
modl <- lm(Species ˜ . , gala)
plot(modl, 1)
We see clear evidence of nonconstant variance. The Box-Cox method reveals that a square-root transformation is a sensible transformation
library(MASS)
boxcox(modl, plotit = TRUE)
modt <- lm(sqrt(Species) ˜ . , gala)
summary(modt)
Call:
lm(formula = sqrt(Species) ˜ ., data = gala)
Residuals:
Min 1Q Median 3Q Max
-4.5572 -1.4969 -0.3031 1.3527 5.2110
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 3.3919243 0.8712678 3.893 0.000690 ***
Area -0.0019718 0.0010199 -1.933 0.065080 .
Elevation 0.0164784 0.0024410 6.751 5.55e-07 ***
Nearest 0.0249326 0.0479495 0.520 0.607844
Scruz -0.0134826 0.0097980 -1.376 0.181509
Adjacent -0.0033669 0.0008051 -4.182 0.000333 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 2.774 on 24 degrees of freedom
Multiple R-squared: 0.7827, Adjusted R-squared: 0.7374
F-statistic: 17.29 on 5 and 24 DF, p-value: 2.874e-07plot(modt, 1)
We achieved this fit at the cost of transforming the response. This makes interpretation more difficult. Furthermore, some of the response values are quite small (single digits) which makes us question the validity of the normal approximation. This model may be adequate, but perhaps we can do better.
modp <- glm(Species ˜ .,family=poisson,gala)
plot(residuals(modp) ˜ predict(modp,type="response"), xlab=expression(hat(mu)), ylab="Deviance residuals")
There are just a few islands with a large predicted number of species while most predicted response values are small. This makes it difficult to see the relationship between the residuals and the fitted values because most of the points are compressed on the left of the display.
plot(residuals(modp) ˜ predict(modp,type="link"),
xlab=expression(hat(eta)),ylab="Deviance residuals")
If we use response residuals
plot(residuals(modp,type="response") ˜ predict(modp,type="link"), xlab=expression(hat(eta)),ylab="Response residuals")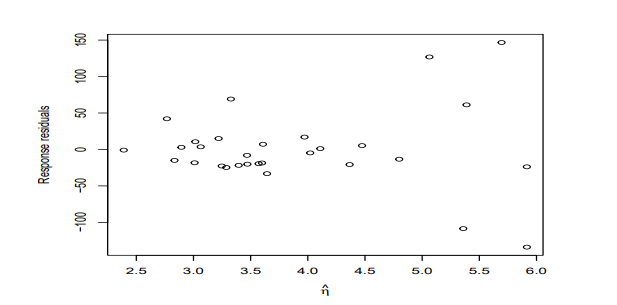
We see a pattern of increasing variation consistent with the Poisson.